
Products
TIP™ Content, Curation & Algorithm Engine
The TIP Database
The Target Informatics Platform's content includes more than 600K high resolution protein structure chains with reliably annotated small molecule binding sites covering every major drug target family including proteases, kinases, phosphatases, phosphodiesterases, nuclear receptors, and GPCRs.
In addition to TIP's high quality structural annotation, it is the only database of its kind that stores all similarity relationships between every sequence, structure, and binding site, making it an incredibly powerful system for structural and comparative proteomics.
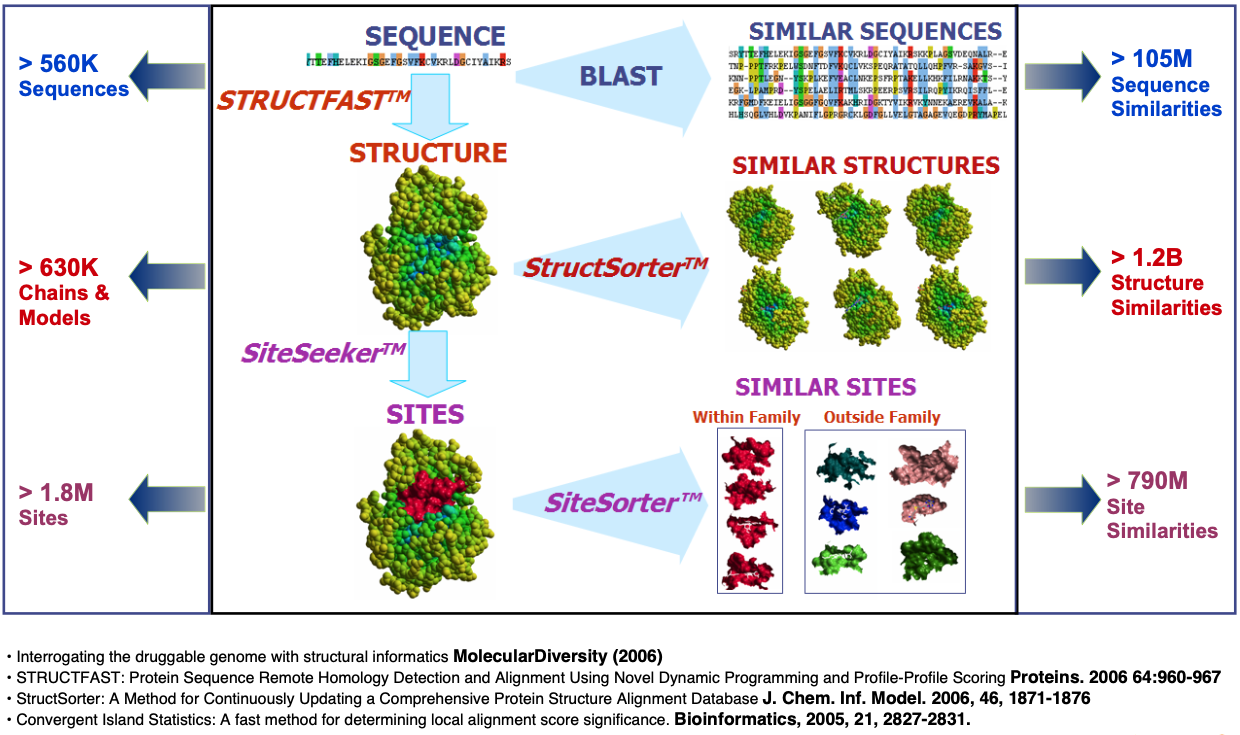
Most importantly, the TIP database automatically and self-consistently updates itself and computes all relevant similarity information when new protein sequences and structures are uploaded into the system. This allows customers who deploy TIP behind their company firewall to upload all of their proprietary sequence and structure data into the system, thereby amplifying the knowledge and maximizing the value that can be derived from their proprietary data.
As an alternative to on-site deployment, secure web-based access to the TIP database can be granted through technology license agreements.
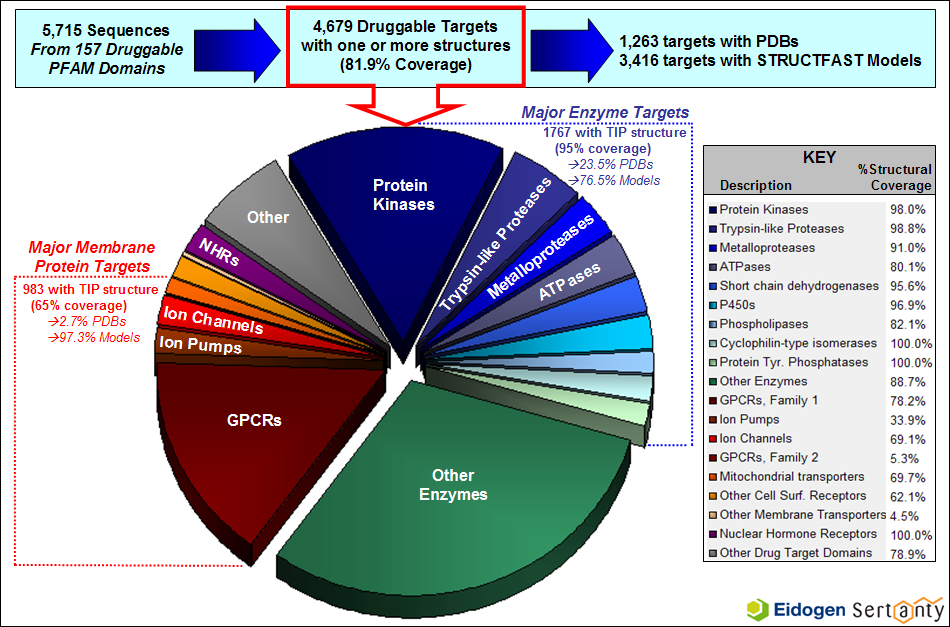
TIP Structural Annotation of Druggable Targets
The TIP database contains a wealth of information about the set of human proteins which have been shown to bind leadlike compounds, commonly referred to as the druggable genome. The figure below highlights TIP's druggable genome coverage along with the amount of structural information available for the major drug target families.
TIP Algorithm Engine
Eidogen's calculation engine for structure determination, binding site annotation, and similarity assessment have been used to generate the largest, most highly integrated database of structural chemogenomics information ever created. Some of the major features of TIP and the corresponding algorithms and software tools responsible for these features are summarized below:
- STRUCTFAST™ - Automated Comparative Modeling
- StructSorter™ - N-by-N Structural Alignments
- SiteSeeker™ - Binding Site Annotation
- SiteSorter™ - N-by-N Binding Site Similarity Assessment
- SLiC™ - Site-Ligand Contact Analysis and Binding Mode SimilarityAssessment
- LigandCross™ - Knowledge-based ligand generation via binding fragment recombination
To find out more about licensing terms and arrange a product demonstration, please contact us at info@eidogen-sertanty.com.